How should patients be monitored for loss of response in the treat-to-target strategy?
How should patients be monitored for loss of response in the treat-to-target strategy?
Mark S. Silverberg, MD
Volume 2 | Issue 6
Runs 1:36
Monitoring for loss of response can include drug monitoring, assessment of biomarkers, and evaluation of the risk of relapse.
Drug monitoring
A proportion of patients with inflammatory bowel disease (IBD) develops adverse events or loses response with tumour necrosis factor-α (TNF-α) inhibitors or thiopurines.(1) Significant clinical data support the utility of therapeutic drug monitoring (TDM) in determining the cause of the problem and identifying an appropriate course of action. Compared with empiric treatment adjustments, TDM provides a logical rationale for dose adjustments or change of agents. The Trough level Adapted infliXImab Treatment (TAXIT) study found patients with Crohn’s disease (CD) randomized to a trough-level dose-adjustment strategy (3–7 μg/mL) had better disease control than those randomized to dose optimization after loss of response, with no increase in cost.(2)
Biologics
Primary lack of response
The reasons for primary lack of response to TNF-α inhibitor therapy are not generally known. However, subtherapeutic drug levels may be one possible explanation, particularly in the setting of acute severe ulcerative colitis (UC). In this setting, a significant inflammatory burden, increased clearance through fecal loss, and accelerated formation of antidrug antibodies (ADAs) all may result in nonresponse to therapy.(1)
Secondary loss of response
Secondary loss of response to TNF-α inhibitor therapy can occur in a variety of clinical contexts, and the appropriate therapeutic action depends on the specific combination of factors. Some general approaches are illustrated in Table 1.(3)
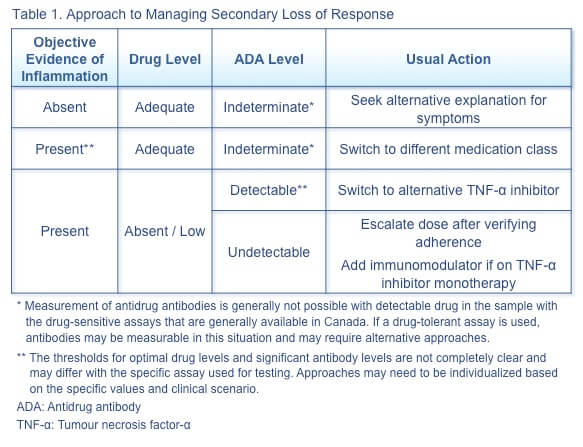
Cross-reactivity between original and biosimilar TNF-α inhibitor
A study to determine the immunogenic comparability between infliximab and CT-P13 (Inflectra) in patients with IBD examined the cross-reactivity with CT-P13 of ADAs to infliximab, using sera from patients with (n=69) and without (n=56) infliximab ADAs. (4) The study found that all infliximab ADA-positive sera cross-reacted with CT-P13, with strongly correlated titres. These sera strongly inhibited binding of infliximab and CT-P13 to TNF-α, demonstrating the functional significance of the ADAs. No infliximab ADA-negative sera cross-reacted with CT-P13. These results suggest that infliximab and CT-P13 have similar immunogencity, immunodominant epitopes, and immunogenic potential. As a result, patients negative for infliximab ADAs can probably be switched to CT-P13, but patients with high-titre infliximab ADAs should probably not be switched to CT-P13.
Adalimumab ADA-positive sera did not react with either infliximab or CT-P13.
Immunomodulators
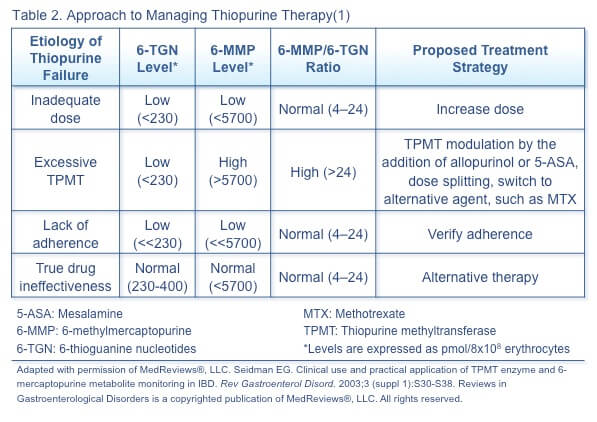
A proportion of patients with IBD may have primary nonresponse to thiopurines, and treatment with these agents may also result in secondary loss of response.(1) The major reasons for thiopurine discontinuation are lack of efficacy and adverse events, both of which may be partially explained by levels of drug metabolites.
Metabolites
The main biologically active metabolite of 6-mercaptopurine (6-MP) and azathioprine is 6-thioguanine nucleotides (6-TGN).(1) This metabolite inhibits lymphocyte proliferation and T-cell apoptosis. Another metabolite, 6-methylmercaptopurine (6-MMP) is associated with hepatotoxicity, although it may also inhibit lymphocyte proliferation. Therapeutic levels of 6-TGN (235–450 pmol/8×108 erythrocytes), are associated with a 5 times greater likelihood of clinical remission than lower levels. As the onset of response to thiopurines is prolonged, determining dosing based on metabolite levels can result in a faster response and reduce costs. Excessive 6-MMP levels (>5,700 pmol/8×108 erythrocytes) increase the risk of hepatotoxicity by 3 times.
Conversion of 6-MP to 6-MMP is catalyzed by thiopurine methyltransferase (TMPT).(1) Measuring TMPT levels at treatment initiation is important, as low or intermediate TMPT activity increases the risk of myelosuppression from accumulation of excessive levels of 6-TGN with standard doses of azathioprine or 6-MP.
Metabolite monitoring
The approach to managing loss of response and adverse events with thiopurines depends on the metabolite levels and ratio (Table 2).(1) Metabolite levels can be measured after 2 to 3 weeks of treatment or dose adjustment and reassessed with loss of response, adverse events, addition of a medication possibly affecting thiopurine metabolism, and twice a year (for routine monitoring and to verify adherence).
Corticosteroids
A oral corticosteroid course can rapidly induce remission in acute moderately-to-severely active IBD.(5) Up to 20% of patients, however, may have corticosteroid-resistant disease. Molecular mechanisms possibly responsible for steroid resistance are altered function of the glucocorticoid receptor, excessive synthesis of proinflammatory cytokines overwhelming the anti-inflammatory capacity of the corticosteroid, and increased drug efflux decreasing plasma levels. Oral corticosteroid failure may respond to intravenous corticosteroid therapy. Alternative therapy with a biologic agent should be considered.
Biomarker assessment
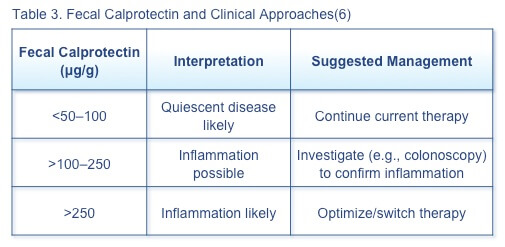
Fecal calprotectin
Fecal calprotectin can discriminate between inflammatory and non-inflammatory gastrointestinal disease, although the role of this biomarker in routine relapse monitoring has not been fully delineated.(6) When the clinician suspects IBD flare, the first bowel movement of the day should be tested for fecal calprotectin. Suggested management of patients with specific fecal calprotectin levels is based on current evidence (Table 3).
A 12-month prospective observational study of 70 patients with UC in endoscopic remission assessing the ability of fecal calprotectin and histologic inflammation to predict relapse found that baseline fecal calprotectin >321 mg/kg predicted disease relapse at 6 and 12 months.(7) Histologic inflammatory activity did not predict relapse, but patients with histologic inflammation had significantly higher fecal calprotectin levels than those without histologic evidence of inflammation. It may be appropriate to follow asymptomatic patients with elevated fecal calprotectin more frequently, as 2 elevated fecal calprotectin levels increase the risk of relapse.
C-reactive protein
In CD, elevated C-reactive protein (CRP) at baseline predicts response to TNF-α inhibitors, and elevation during therapy is an independent predictor of relapse.(8) Elevated CRP at baseline in patients with severe UC predicts failure of medical therapy. CRP normalization has been associated with remission in patients with IBD, and failure of normalization with treatment should prompt additional evaluation.
Prediction of relapse and severe disease
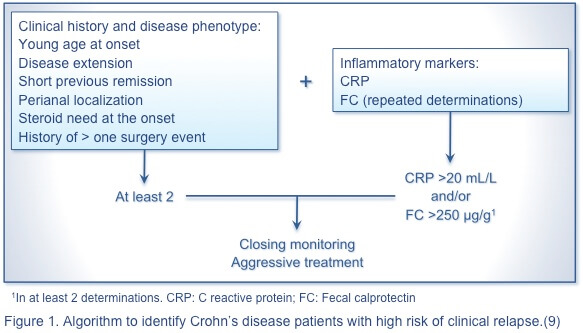
To date, the occurrence of disease flares in patients with IBD has not been predictable.(9) An array of predictors of severe disease and disease relapse have been identified. These include parameters such as endoscopic activity, inflammatory markers, laboratory parameters, fecal markers, and tests still primarily in the research realm such as cytokines, composition of the gut microbiota, intestinal permeability, NOD2 genetic polymorphisms, and immune-mediated antibodies, such as pANCA and ASCA.(9,10)
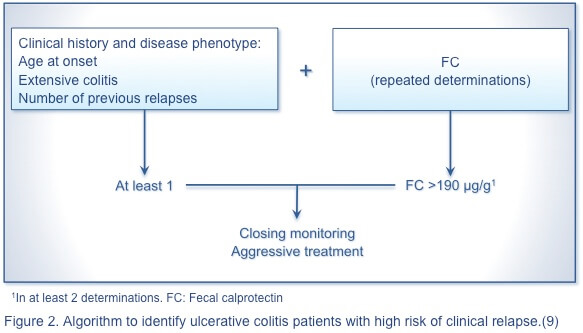
Nevertheless, no single marker or panel of markers reliably predicts disease relapse in UC or CD. The following algorithmic approach is proposed to identify patients with CD (Figure 1) and those with UC (Figure 2) with a high risk of clinical relapse.(9) A simple, rapid, economical, and noninvasive predictor of severe disease is needed to advance the management of IBD.
References
- Kopylov U, Ben-Horin S, Seidman E. Therapeutic drug monitoring in inflammatory bowel disease. Ann Gastroenterol. 2014;27(4):304–12.
- Vande Casteele N, Compernolle G, Ballet V, et al. Individualised infliximab treatment using therapeutic drug monitoring: a prospective controlled trough level adapted infliXImab treatment (TAXIT) trial. J Crohns Colitis. 2012;6:S6.
- Khanna R, Sattin BD, Afif W, Benchimol EI, Bernard EJ, Bitton A, et al. Review article: a clinician’s guide for therapeutic drug monitoring of infliximab in inflammatory bowel disease. Aliment Pharmacol Ther. 2013;38(5):447-59.
- Ben-Horin S, Yavzori M, Benhar I, et al. Cross-immunogenicity: antibodies to infliximab in Remicade-treated patients with IBD similarly recognise the biosimilar Remsima. Gut. 2016;65(7):1132–
- Quetglas EG, Armuzzi A, Wigge S, et al. Review article: The pharmacokinetics and pharmacodynamics of drugs used in inflammatory bowel disease treatment. Eur J Clin Pharmacol. 2015;71:773–99.
- Bressler B, Panaccione R, Fedorak RN, et al. Clinicians’ guide to the use of fecal calprotectin to identify and monitor disease activity in inflammatory bowel disease. Can J Gastroenterol Hepatol. 2015;29(7):369–
- Theede K, Holck S, Ibsen P, et al. Fecal calprotectin predicts relapse and histological mucosal healing in ulcerative colitis. Inflamm Bowel Dis. 2016;22:1042–8.
- Murdoch T, O’Donnell S, Silverberg MS, et al. Biomarkers as potential treatment targets in inflammatory bowel disease: A systematic review. Can J Gastroenterol Hepatol. 2015;29(4):203–
- Liverani E, Scaioli E, Digby RJ, et al. How to predict clinical relapse in inflammatory bowel disease patients. World J Gastroenterol. 2016;22(3):1017–
- Musci JO, Cornish JS, Dabritz J. Utility of surrogate markers for the prediction of relapses in inflammatory bowel diseases. J Gastroenterol. 2016;51:531–47.
Special Edition IBD Dialogue 2016 Volume 02: Treat-to-Target in IBD is made possible by an unrestricted educational grant from…
![]()